[陳柏仰] BSImp:估算鄰近甲基化模式出現的機率以增補甲基化資料
發稿時間: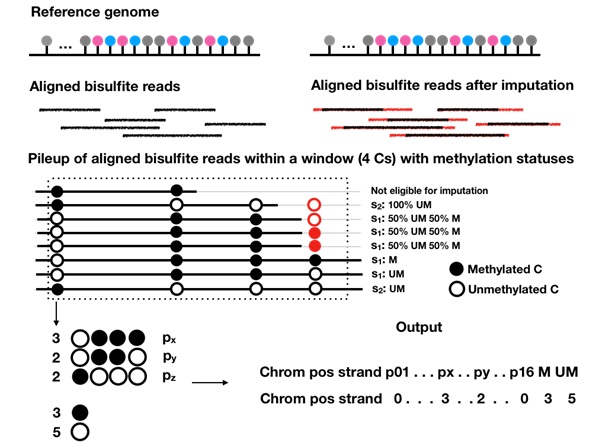
圖:甲基化缺值的估算流程。首先將定序資料與參考基因組對齊,之後將整個基因組切成相同數量位點的區間(這裡例子是4個胞嘧啶位點)分別估算缺值。藉由分析區間內序列的甲基化樣式估算缺值。輸出的資料包括染色體、位置、方向、區間內每種甲基化樣式的數量、以及甲基化和未甲基化胞嘧啶的數量。
DNA 甲基化是研究最多的表觀遺傳修飾之一,可影響包括基因的調控以及老化的進程。藉由亞硫酸鹽定序 (BS-seq) 及酵素轉換甲基定序(EM-seq)方式可以得到單鹼基分辨率的甲基化資訊。每條序列的甲基化狀態反映單一細胞的甲基化樣式(methylation patterns),而分析特定區域眾多甲基化樣式可以反應細胞群內的異質性(cellular heterogeneity),而這些異質性可以用來預測發育和疾病。因此,偵測甲基化異質性(methylation heterogeneity)具有發現細胞早期變化的潛力。目前已開發的甲基化異質性偵測工具是分析多個連續 CpG 形成的甲基化樣式,但次世代定序所得到的短序列通常無法得到完整的甲基化樣式,使得可分析的數據非常有限。到目前為止只有一個工具可以用來估算甲基化樣式,但是此工具需要使用大量的數據來訓練,且也無法分析整個基因組。因此,我們開發了一種基於機率的插補方法,使用來自鄰近位點的甲基化資訊來快速填補不完整的甲基化樣式。我們的工具可以增加百分之十五以上的區域用於評估甲基化異質性,並具有高度的準確性。此程式可輸出全基因組的甲基化樣式,直接用於評估甲基化程度以及甲基化異質性。我們的方法可以準確的估計甲基化程度,並且產生合理的覆蓋範圍作為甲基化異質性的分析,這對於利用分析細胞群內的甲基化異質性變化來評估生長和疾病具有重要意義。