[Pao-Yang Chen] BSImp: imputing partially observed methylation patterns for evaluating methylation heterogeneity
POST: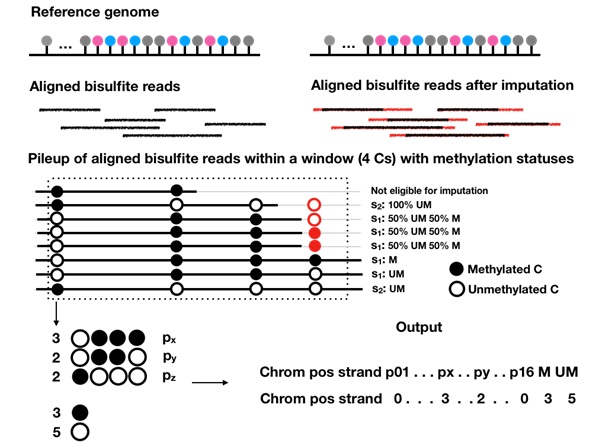
Figure: Schematic representation of the output of BSImp. First the reads are aligned to the reference genome, given any window of specified number of cytosines (here is 4), methylation statuses are extracted and missing values imputed if valid, then methylation profiled as a vector including chromosome, position, strand, the copy numbers of all possible methylation patterns starting at the position, and number of methylated and unmethylated cytosines.
DNA methylation is one of the most studied epigenetic modifications that has applications ranging from transcriptional regulation to aging, and can be assessed by bisulfite sequencing (BS-seq) or enzymatic methyl sequencing (EM-seq) at single base-pair resolution. The permutations of methylation statuses given by aligned reads reflect the methylation patterns of individual cells. These patterns at specific genomic locations are sought to be indicative of cellular heterogeneity within a cellular population, which are predictive of developments and diseases; therefore, methylation heterogeneity has potentials in early detection of these changes. Computational methods have been developed to assess methylation heterogeneity using methylation patterns formed by four consecutive CpGs, but the nature of shotgun sequencing often give partially observed patterns, which makes very limited data available for downstream analysis. While many programs are developed to impute genome-wide methylation levels, currently there is only one method developed for recovering partially observed methylation patterns; however, the program needs lots of data to train and cannot be used directly; therefore, we developed a probabilistic-based imputation method that uses information from neighbouring sites to recover partially observed methylation patterns speedily. It is demonstrated to allow for the evaluation of methylation heterogeneity at 15% more regions genome-wide with high accuracy for data with moderate depth. To make it more user-friendly we also provide a computational pipeline for genome-screening, which can be used in both evaluating methylation levels and profiling methylation patterns genomewide for all cytosine contexts, which is the first of its kind. Our method allows for accurate estimation of methylation levels and makes evaluating methylation heterogeneity available for much more data with reasonable coverage, which has important implications in using methylation heterogeneity for monitoring changes within the cellular populations that were impossible to detect for the assessment of development and diseases.