[Chuan-Chih Hsu] Suspension Trapping-Based Sample Preparation Workflow for In-Depth Plant Phosphoproteomics
POST: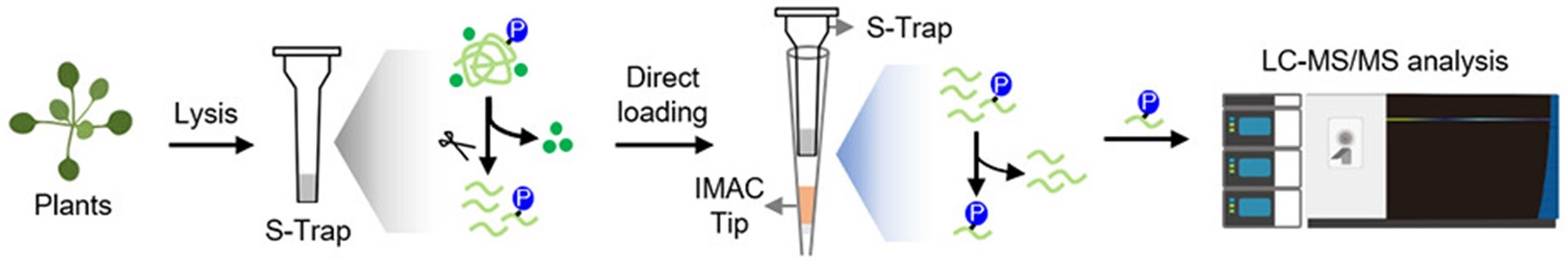
Experimental design of the tandem S-Trap-IMAC workflow for plant phosphoproteomics. Interfering contaminants in the plant lysate are removed by washing the S-Trap, and proteins are digested within the S-Trap. Phosphopeptides are enriched using an Fe-IMAC tip via a direct loading strategy and analyzed using a Fusion Lumos mass spectrometer in data-dependent acquisition mode.
Plant phosphoproteomics provides a global view of phosphorylation-mediated signaling in plants; however, it demands high-throughput methods with sensitive detection and accurate quantification. Despite the widespread use of protein precipitation for removing contaminants and improving sample purity, it limits the sensitivity and throughput of plant phosphoproteomic analysis. The multiple handling steps involved in protein precipitation lead to sample loss and process variability.
In this context, the Proteomics Core Lab developed an approach based on suspension trapping (S-Trap), termed tandem S-Trap-IMAC, by integrating an S-Trap micro-column with a Fe-IMAC tip. Compared with a precipitation-based workflow, the tandem S-Trap-IMAC method deepened the coverage of the Arabidopsis phosphoproteome by more than 30%, with improved number of multiply phosphorylated peptides, quantification accuracy, and short sample processing time.
The tandem S-Trap-IMAC method was employed to study abscisic acid (ABA) signaling in Arabidopsis seedlings. The results thus showed that a significant proportion of the phosphopeptides induced by ABA are multiply phosphorylated peptides, indicating their importance in early ABA signaling and quantified several key phosphorylation sites on core ABA signaling components across four time points.
The first author, Chin-Wen Chen, is a research assistant in the Proteomics Core Lab. The study is published in the journal of Analytical Chemistry.