[Pao-Yang Chen] There’s More to It: Uncovering Genomewide DNA Methylation Heterogeneity
POST: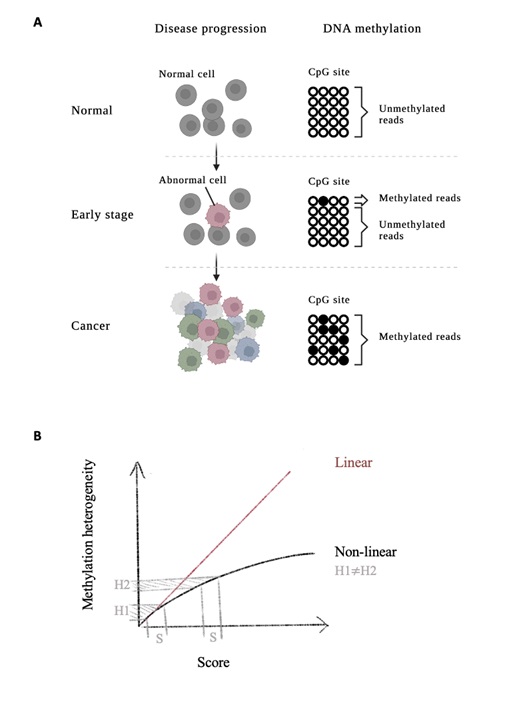
Estimating DNA methylation heterogeneity (A) DNA methylation heterogeneity for monitoring disease progression (B) Linear and nonlinear scores in measuring DNA methylation heterogeneity.
DNA methylation heterogeneity is a metric that describes how variable the methylation statuses are between cells within a population. This metric has been shown to elucidate biological processes, such as disease progression and cell development. Several computational tools have been developed in the recent years to measure methylation heterogeneity in pooled cell samples. This is radical as it means that all existing pooled cell datasets have a chance to be further dissected for heterogeneity without the need for single cell isolation. Depending on their frameworks, these tools have their individual strengths and weaknesses, but they also share significant limitations. For example, we noticed that the scoring systems used do not linearly describe heterogeneity, which prevent direct comparison of heterogeneity across loci. In this editorial, we highlight the importance of reliably capture all cases of methylation heterogeneity. We look forward for methylation heterogeneity to shed light on questions that cannot be answered with averaged methylation values alone, such as deciphering the role of heterogeneity in transcriptional regulation. In the future, it is expected that these methods will be extended to cover non-CG contexts which are dominant in plants, as well as to be used to profile methylation heterogeneity of highly complex genomic regions with the help of long-read technology.
This is an invited editorial published in Epigenomics. The leading authors are Jenny HueyTyng Lee and Pei-Yu Lin.