[Pao-Yang Chen] MethGET: web-based bioinformatics software for correlating genome-wide DNA methylation and gene expression
POST: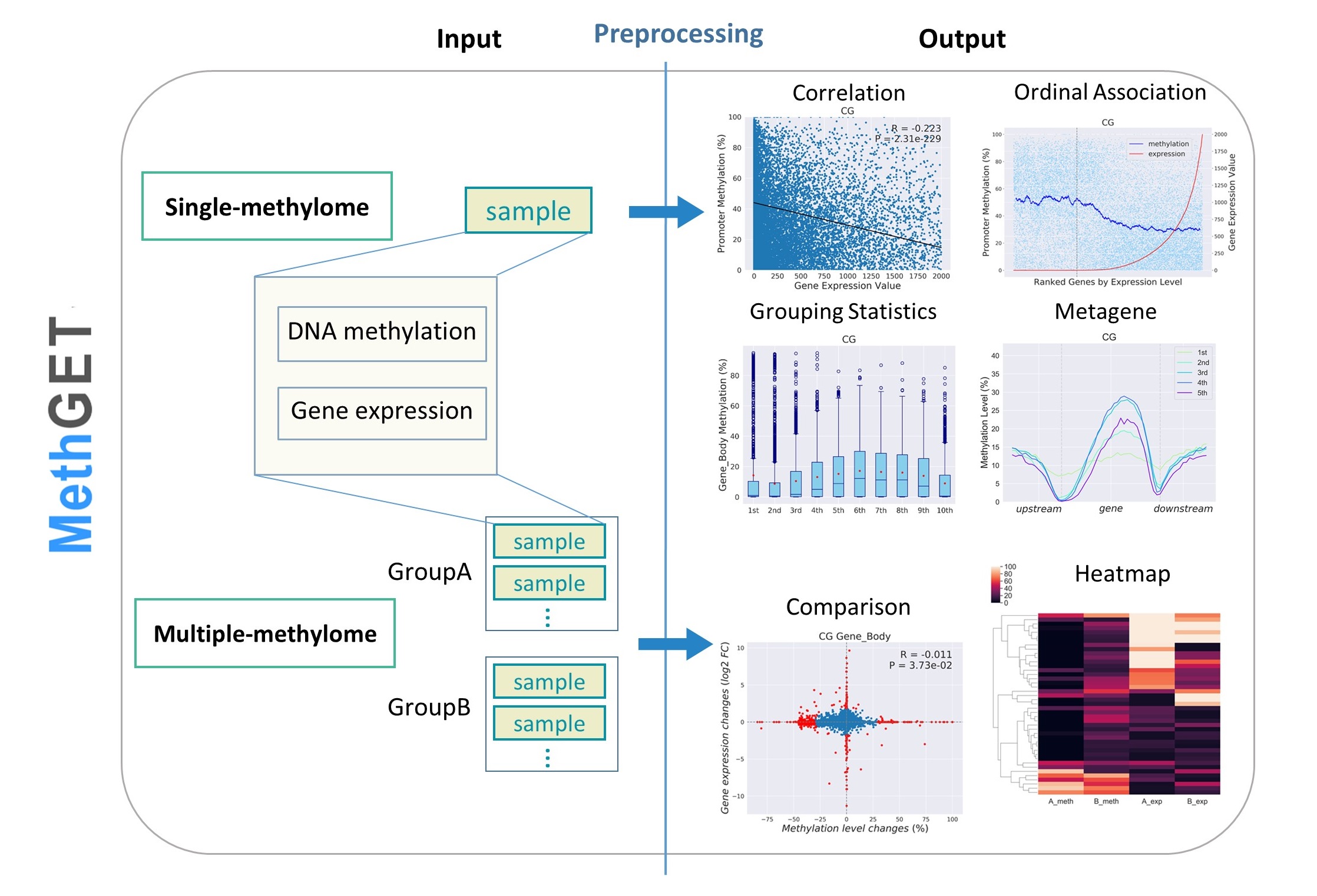
Figure. Schematic diagram of MethGET. The diagram shows the inputs and outputs of single-methylome analyses and multiple-methylome analyses.
Background
DNA methylation is a major epigenetic modification involved in regulating gene expression. The effects of DNA methylation on gene expression differ by genomic location and vary across kingdoms, species and environmental conditions. To identify the functional regulatory roles of DNA methylation, the correlation between DNA methylation changes and alterations in gene expression is crucial. With the advance of next-generation sequencing, genome-wide methylation and gene expression profiling have become feasible. Current bioinformatics tools for investigating such correlation are designed to the assessment of DNA methylation at CG sites. The correlation of non-CG methylation and gene expression is very limited. Some bioinformatics databases allow correlation analysis, but they are limited to specific genomes such as that of humans and do not allow user-provided data.
Results
Here, we developed a bioinformatics web tool, MethGET (Methylation and Gene Expression Teller), that is specialized to analyse the association between genome-wide DNA methylation and gene expression. MethGET is the first web tool to which users can supply their own data from any genome. It is also the tool that correlates gene expression with CG, CHG, and CHH methylation based on whole-genome bisulfite sequencing data. MethGET not only reveals the correlation within an individual sample (single-methylome) but also performs comparisons between two groups of samples (multiple-methylomes). For single-methylome analyses, MethGET provides Pearson correlations and ordinal associations to investigate the relationship between DNA methylation and gene expression. It also groups genes with different gene expression levels to view the methylation distribution at specific genomic regions. Multiple-methylome analyses include comparative analyses and heatmap representations between two groups. These functions enable the detailed investigation of the role of DNA methylation in gene regulation. Additionally, we applied MethGET to rice regeneration data and discovered that CHH methylation in the gene body region may play a role in the tissue culture process, which demonstrates the capability of MethGET for use in epigenomic research.
Conclusions
MethGET is a Python software that correlates DNA methylation and gene expression. Its user-friendly web interface facilitates the investigation of gene expression regulation by DNA methylation (https://paoyang.ipmb.sinica.edu.tw/methget). The stand-alone version and source codes are available on GitHub at https://github.com/Jason-Teng/MethGET.
* Chin-Sheng Teng is from IPMB Summer Undergraduate Internship Program.