[Chih-Horng Kuo] Analysis of Xylella genomes and virulence genes
POST: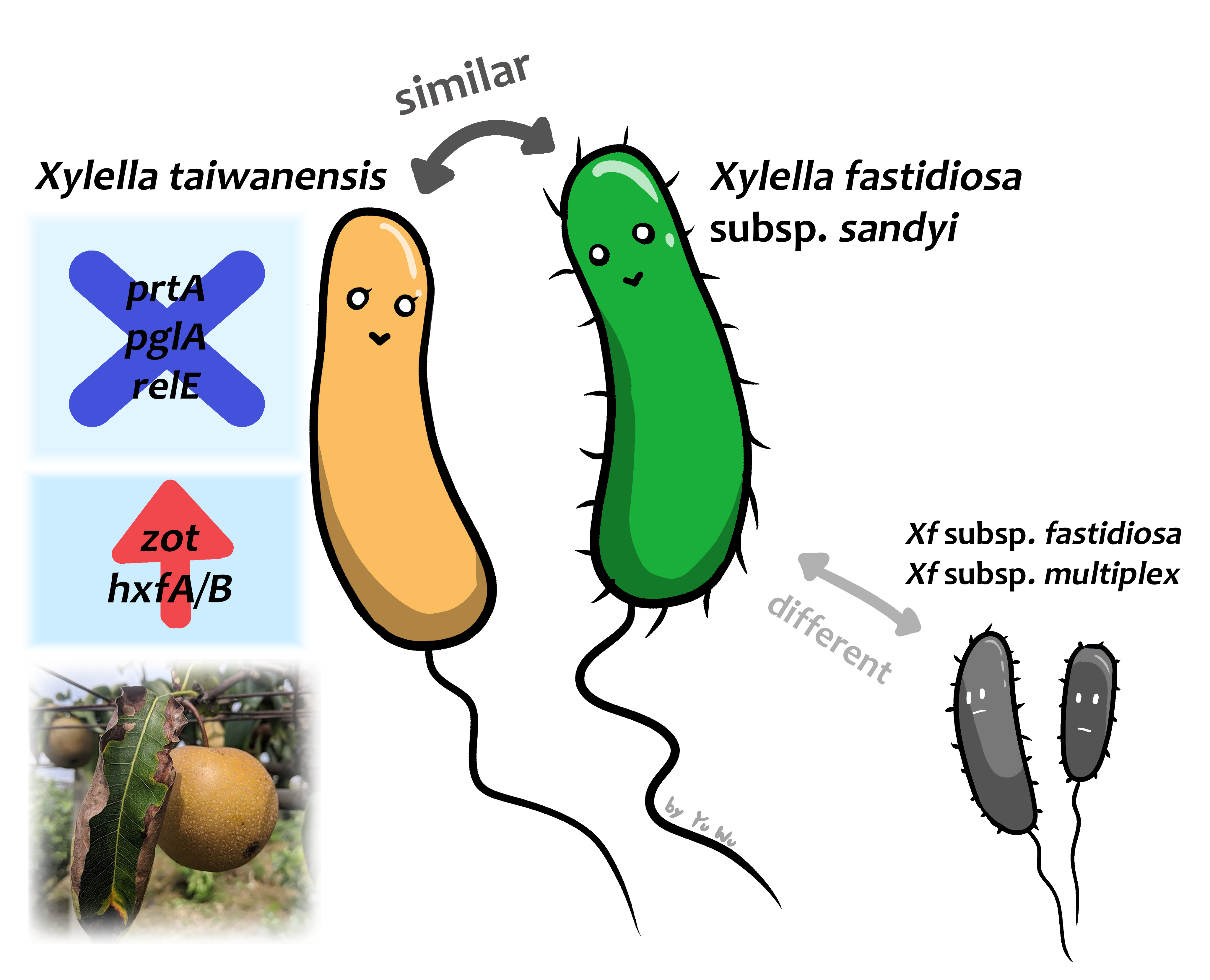
The bacterium Xylella fastidiosa is a plant pathogen transmitted by insect vectors. This species contains multiple subspecies, such as X. fastidiosa subsp. fastidiosa that causes Pierce's disease in grapevines, X. fastidiosa subsp. pauca that causes citrus variegated chlorosis disease, and X. fastidiosa subsp. sandyi that causes oleander leaf scorch disease, which represent major threats to agriculture and ecology in America and Europe. Due to their importance, these pathogens have received much research attention. Particularly, genomics tools provide a powerful approach to study these bacteria that are difficult to culture. However, previous works have focused on the characterization of genetic diversity and evolution at the levels of between- or within-subspecies, whereas comparative analysis at the between-species or high levels was lacking. This limitation was due to the fact that X. fastidiosa has been the only species described in this genus for decades, such that there is no appropriate data set for between-species comparison. At higher levels, Xanthomonas is the most closely related group. However, the genomic divergence between Xylella and Xanthomonas is very high in terms of chromosomal organization and sequence variation, and the exact phylogenetic relationships remain controversial, making the extraction of biological insights from such comparison difficult.
For Xylella-related research in Taiwan, pear cultivar Hengshan grown at elevations below 1,200 m suffered pear leaf scorch disease since around 1980 and the disease incidence is in the range of 5-43%. The causative agent of this disease was confirmed in 1990 and identified as X. fastidiosa. However, later works confirmed that this bacterium is a novel species endemic to Taiwan, and it was formally described as Xylella taiwanensis in 2016, thus becoming the second species of this important genus. To take advantage of this valuable biological resource for in-depth investigation of Xylella genomic variation, Dr. Chih-Horng Kuo in our institute collaborated with Drs. Chi-Wei Tsai (Department of Entomology, National Taiwan University) and Chiou-Chu Su (Taiwan Agricultural Chemicals and Toxic Substances Research Institute, Council of Agriculture) to determine the complete genome sequence of the type strain of X. taiwanensis for comparative analysis with other Xanthomonadaceae representatives. The results indicated that Xylella evolved from within Xanthomonas through genome reduction and is more closely related to the clade represented by Xanthomonas campestris, while more distantly related to the clade represented by Xanthomonas albilineans. Detailed phylogenetic analysis within the genus Xylella further confirmed that X. fastidiosa subsp. pauca is the basal lineage that has the earliest divergence from other subspecies. For comparison of overall gene content, X. taiwanensis is most similar to X. fastidiosa subsp. sandyi and both have a narrow host range. For detailed examination of virulence gene content among different Xylella lineages, those involved in secretion and metabolism are the most conserved ones, while those related to adhesins, hydrolytic enzymes, and toxin-antitoxin systems are highly variable. Those virulence genes with high levels of conservation or variation may be affected most by natural selection, thus are promising candidates for future molecular genetics studies for further understanding of the molecular mechanisms for plant-microbe interactions.
Weng LW#, Lin YC#, Su CC, Huang CT, Cho ST, Chen AP, Chou SJ, Tsai CW*, Kuo CH* (2021) Complete genome sequence of Xylella taiwanensis and comparative analysis of virulence gene content with Xylella fastidiosa. Frontiers in Microbiology 12:684092. DOI: 10.3389/fmicb.2021.684092 [bioRxiv preprint DOI: 10.1101/2021.03.08.434500] #Equal contribution.